
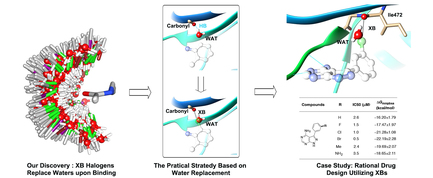
Brief introduction: Halogen bonds (XBs) are increasingly attracting attention in the field of structure-based drug design, limited utilization of XBs in structure-based drug design has stimulated efforts to develop more effective computational guidance. Here, we analyzed the binding-site water properties of crystal structures with characterized XBs between ligand halogen atoms and protein backbone carbonyls and, thus, found that halogen atoms involved in XBs frequently replace binding-site waters upon ligand binding. Moreover, we observed that the preferential directionality of XBs aligned well with the orientations of these replaced waters, and these replaced waters exhibited differential energetic characteristics. Combining with the geometrical characteristics of XBs, we proposed a practical strategy for rational drug design utilizing XBs. Then, we explored XBs between the scaffold of ibrutinib and its kinase target protein BTK based on the strategy, and the XBs were validated by systematic structure-activity relationship studies and free energy calculations. Our studies provided a practical approach for rational drug design utilizing XBs with protein backbone carbonyl groups.