
Abstract
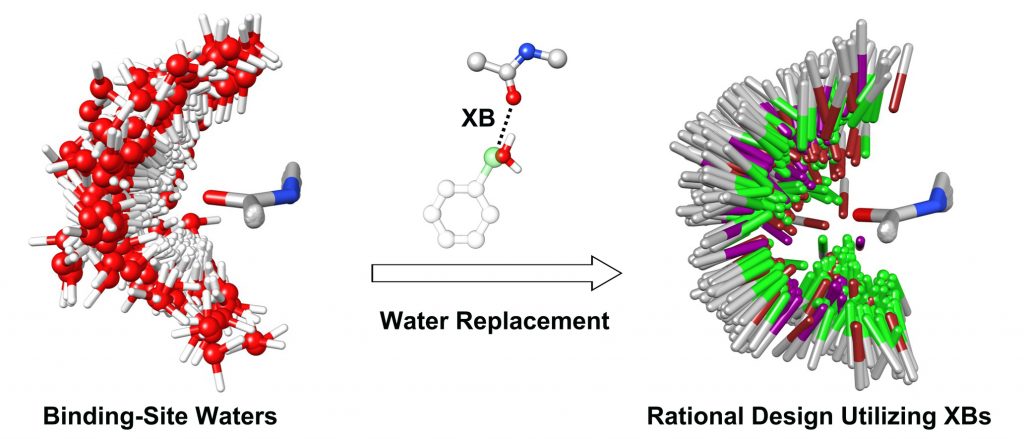
Halogen bond interaction between a protein electronegative atom and a ligand halogen atom is increasingly attracting attention in the field of structure-based drug design. Nevertheless, gaps in understanding make it desirable to better examine the role of forces governing the formation of favorable halogen bond interactions, and the development of effective and efficient computational approaches to “design in” favorable halogen bond interactions in lead optimization process are warranted. Here, we analyzed the binding-site water properties of crystal structures with characterized halogen bond interactions between ligand halogen atoms and protein backbone carbonyl groups and, thus, found that halogen atoms involved in halogen bond interactions frequently replace calculated binding-site waters upon ligand binding. Moreover, we observed that the preferential directionality of halogen bond interactions aligns well with the orientations of these replaced waters, and these replaced waters exhibited differential energetic characteristics as compared to waters that are displaced by halogen atoms that do not form halogen bond interactions. Our discovery that replacement of calculated binding-site waters contributes to the formation of favorable halogen bond interactions suggests a practical approach for rational drug design utilizing halogen bond interactions with protein backbone carbonyl groups.

Replacement of Protein Binding-Site Waters Contributes to Favorable Halogen Bond Interactions.
Wang Y1,2, Fu Q2,3, Zhou Y2, Du Y1, Huang N2,4.
1.School of Pharmaceutical Science & Technology , Tianjin University , Tianjin 300072 , China.
2.National Institute of Biological Sciences , Beijing, No. 7 Science Park Road, Zhongguancun Life Science Park , Beijing 102206 , China.
3.College of Biological Sciences , China Agricultural University , Beijing 100193 , China.
4.Tsinghua Institute of Multidisciplinary Biomedical Research , Tsinghua University , Beijing 102206 , China.