
Incorporating replacement free energy of binding-site waters in molecular docking
Hanzi Sun1,2,Lifeng Zhao2,Shiming Peng2,Niu Huang1,2,*
1. College of Life Sciences, Beijing Normal University, Beijing, China
2. National Institute of Biological Sciences, Beijing, Zhongguancun Life Science Park, Beijing, China
*Correspondence to: Niu Huang, National Institute of Biological Sciences, Beijing, No. 7 Science Park Road, Zhongguancun Life Science Park, Beijing 102206, China. E-mail: huangniu#nibs.ac.cn
ABSTRACT
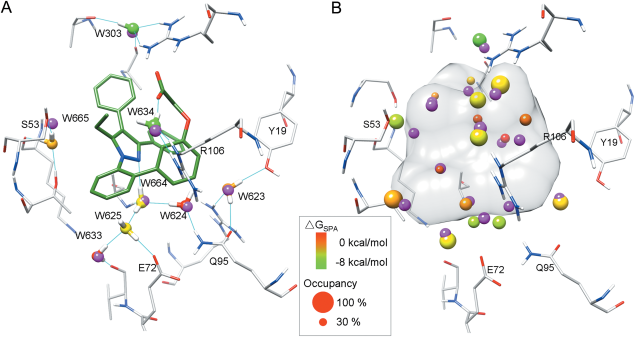
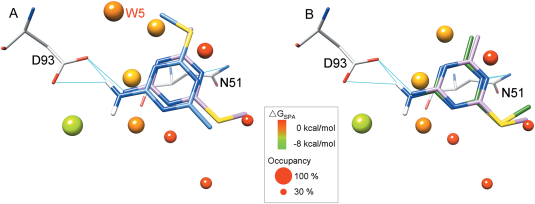
Binding-site water molecules play a crucial role in protein-ligand recognition, either being displaced upon ligand binding or forming water bridges to stabilize the complex. However, rigorously treating explicit binding-site waters is challenging in molecular docking, which requires to fully sample ensembles of waters and to consider the free energy cost of replacing waters. Here, we describe a method to incorporate structural and energetic properties of binding-site waters into molecular docking. We first developed a solvent property analysis (SPA) program to compute the replacement free energies of binding-site water molecules by post-processing molecular dynamics trajectories obtained from ligand-free protein structure simulation in explicit water. Next, we implemented a distance-dependent scoring term into DOCK scoring function to take account of the water replacement free energy cost upon ligand binding. We assessed this approach in protein targets containing important binding-site waters, and we demonstrated that our approach is reliable in reproducing the crystal binding geometries of protein-ligand-water complexes, as well as moderately improving the ligand docking enrichment performance. In addition, SPA program (free available to academic users upon request) may be applied in identifying hot-spot binding-site residues and structure-based lead optimization.Proteins 2014. © 2014 Wiley Periodicals, Inc.