
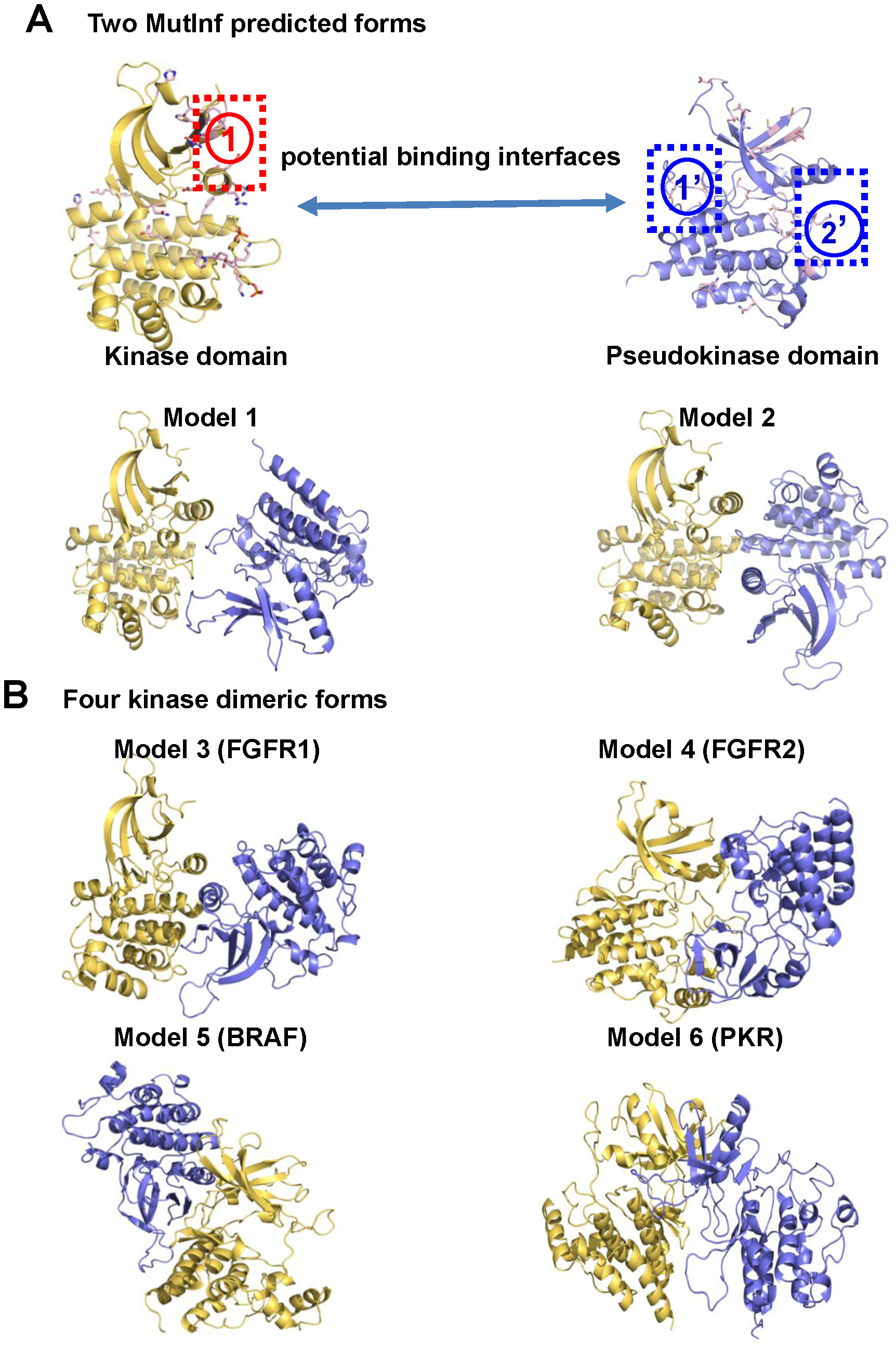
The Janus Kinase 2 (JAK2) plays essential roles in transmitting signals from multiple cytokine receptors, and constitutive activation of JAK2 results in hematopoietic disorders and oncogenesis. JAK2 kinase activity is negatively regulated by its pseudokinase domain (JH2), where the gain-of-function mutation V617F that causes myeloproliferative neoplasms resides. In the absence of a crystal structure of full-length JAK2, how JH2 inhibits the kinase domain (JH1), and how V617F hyperactivates JAK2 remain elusive. We modeled the JAK2 JH1–JH2 complex structure using a novel informatics-guided protein-protein docking strategy. A detailed JAK2 JH2-mediated auto-inhibition mechanism is proposed, where JH2 traps the activation loop of JH1 in an inactive conformation and blocks the movement of kinase αC helix through critical hydrophobic contacts and extensive electrostatic interactions. These stabilizing interactions are less favorable in JAK2-V617F. Notably, several predicted binding interfacial residues in JH2 were confirmed to hyperactivate JAK2 kinase activity in site-directed mutagenesis and BaF3/EpoR cell transformation studies. Although there may exist other JH2-mediated mechanisms to control JH1, our JH1–JH2 structural model represents a verifiable working hypothesis for further experimental studies to elucidate the role of JH2 in regulating JAK2 in both normal and pathological settings.